| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
Nefropatiile tubulare
Definirea nefropatiilor si sindroamelor tubulare
O serie de conditii patologice variate, ereditare sau dobandite, pot altera functiile de transport tubular, productia tubulara de hormoni sau substante biologic active, respectiv raspunsul tubular la acestea, realizand sindromul nefropatiei tubulare, cu evolutie acuta sau cronica.
Nefropatiile tubulare acute sunt dobandite, se caracterizeaza functional prin alterarea tuturor functiilor tubulare si multe se manifesta ca injurie renala acuta.[¹] Este remarcabila rapiditatea cu care se instaleaza si extinderea leziunilor tubulare.
Se pot asocia cu leziuni inflamatorii acute interstitiale exprimate prin infiltrat inflamator format din plasmocite, limfocite, eozinofile si polimorfonucleare, dispuse difuz sau nodular, motiv pentru care sunt cunoscute si sub denumirea de nefropatii tubulointerstitiale acute (NTIA).[²]
Sindromul nefropatiilor tubulare cronice este reprezentat de boli si sindroame cu transmitere genetica (ereditare) sau dobandite, caracterizate structural prin alterari histologice sau numai biochimice, moleculare ce afecteaza predominant, primitiv sau secundar, sistemul tubular al nefronului si functional prin alterarea uneia sau a mai multor functii tubulare cu manifestari de insuficienta tubulara proximala, distala sau proximo-distala.[¹] Spre deosebire de nefropatiile tubulare acute, nu se insotesc de edem interstitial, infiltrat polimorfonuclear si eozinofilie.[²]
Nefropatii tubulare cronice
Clasificare
In functie de modalitatea afectarii tubulare, tubulopatiile cronice pot fi clasificate in: primitive sau secundare.
Tubulopatii cronice primitive
In tubulopatiile cronice primitive afectarea este primitiv tubulara. Cele mai multe dintre ele sunt boli sau sindroame ereditare. Factorii genetici pot modifica functiile tubulare prin doua mecanisme:
determinand o anomalie primitiva a functiei tubulare (ex. diabetul renal);
mediind un efect toxic asupra tubului renal, secundar acumularii de substante din diferite metabolisme extrarenale (ex. cistinoza).
Ambele mecanisme determina un tablou simtomatologic comun, functional si urinar.
Teoretic, o tubulopatie cronica ereditara poate aparea in:
-defect al interactiunii dintre substrat si receptorul membranar;
-defect in producerea sau eliberarea energiei necesare transportului;
-limitarea gradientului procesului de transport datorita cresterii pool-ului intracelular de substanta;
-eflux excesiv in lumen al substantei reabsorbite, producand reducerea seabsorbtiei nete sau chiar secretiei aparente;
-influx excesiv al substantei secretate, producand scaderea secretiei nete sau chiar reabsorbtie aparenta;
-deficit de sinteza sau eliberare a hormonilor si substantelor biologic-active de catre celulele tubulare;
-absenta raspunsului tubular renal la hormoni, datorat fie unui defect al interactiunii hormon-receptor, fie unei anomalii post-receptor. [¹]
Tubulopatii cronice secundare
In tubulopatiile cronice secundare atingerea tubulara este secundara unor boli renale sau extrarenale, ereditare sau dobandite.[¹]
In functie de etiologie, tubulopatiile se clasifica in:
Primitive
Diabet renal ereditar
a) Simplu
Diabet insipid nefrogen
Diabet renal al Na,K, Ca, Mg
Diabet renal fosfaturic
Diabet renal glicozuric
Diabet renal aminoaciduric
b) Complex
Diabet dublu ( gluco-aminoaciduric, gluco-fosfaturic, amino-fosfaturic)
Diabet complex ( Sindrom Toni-Debre-Fanconi, boala Hartnup)
Acidoze tubulare renale
Secundare
Boli ereditare ale metabolismului
Boli dobandite:
a. Intoxicatii: metale grele, sulfamide, tetraciclina alterata, vitamina D
b. Carenta de vitamina D (rahitismul comun)
c. Dezechilibre hidro-electrolitice (deshidratari, hipercalcemie, depletie de potasiu)
d. Neoplazii
e. Boli autoimune: LED, sindrom Sjogren
f. Disgamaglobulinemii (mielom, amiloidoza)
g. Nefropatii: interstitiale, sindrom nefrotic
h. Uropatii: congenitale sau dobandite.¹
In functie de segmentul afectat nefropatiile tubulare cronice mai pot fi clasificate in: proximale, distale si proximo-distale [3].
Tubulopatii proximale cronice
Diabetul renal glicozuric (Glicozuria normoglicemica ereditara)
Definitie
Diabetul renal glicozuric (DRG) este o tubulopatie ereditara cu transmitere autozomal recesiva, caracterizata prin glicozurie in absenta hiperglicemiei. Homozigotii pot prezenta glicozurie > 60 g/dl, pierdere de sodiu urinar, depletie de volum, niveluri crescute de aldosteron si renina plasmatica.[4] Unele cazuri au fost asociate cu aminoaciduria selectiva.[5]
Fiziopatologie
Fiziologic, glucoza este filtrata glomerular si se afla in concentratie egala cu cea din plasma; este reabsorbita la nivelul tubului renal proximal printr-un proces de transport dependent de Na. Capacitatea de reabsorbtie tubulara depaseste concentratia normala a glicemiei. Pragul urinar de eliminare a glucozei este depasit la o valoare a glicemiei de >200 mg/dl iar capacitatea maxima de reabsorbtie este depasita la o incarcatura filtrata in jurul valorii de 325 mg/min/1.73m², aceasta valoare fiind definita ca maximum tubular pentru glucoza (TmG).
F. Reubi a descris doua tipuri de diabet renal glicozuric:
-Tipul A, caracterizat atat prin reducerea pragului renal al glucozei (TG) cat si a capacitatii maxime de reabsorbtie a glucozei (TmG). Acesti pacienti prezinta, in cursul perfuziei cu glucoza, glicozurie importanta la concentratii relativ mici, iar daca se creste progresiv glicemia, cantitatea de glucoza reabsorbita atinge repede un platou pe care nu-l poate depasi.
-Tipul B, caracterizat prin reducerea pragului renal al glucozei, dar cu TmG normal. Pacientii prezinta, in cursul perfuziei cu glucoza, glicozurie moderata la concentratii plasmatice joase, dar reabsorbtia este normala la supraincarcarea cu glucoza [6].
Clinic
Bolnavul este de obicei asimptomatic si glicozuria este descoperita intamplator cu ocazia unui examen de rutina. Cand glicozuria este importanta apar semne clinice de hipoglicemie, poliurie, polifagie.
Paraclinic
-Glicozurie permanenta
-Glicemie normala, care nu depaseste 170 mg% la proba hiperglicemiei provocate.
-Absenta proteinuriei, aminoaciduriei, sediment urinar normal.
Diagnosticul pozitiv se formeaza pe baza urmatoarelor date:
1.Ancheta familiala
2.Glicozurie permanenta
3. Glicemie a jeun normala; postprandial nu creste peste 150 mg%
4. Glicemie mai mica de 170 mg% la testul de hiperglicemie provocata.
5. Scaderea TmG si TG
Diagnostic diferential
Glicozuriile renale din sindromul Fanconi
Glicozuriile normoglicemice din nefropatiile organice
Glicozuria normoglicemica din boala Wilson
Glicozurii hiperglicemice (cel mai frecvent in DZ) [6]
Tratament
Nu exista tratament etiopatogenic. Singura masura terapeutica este prescrierea unei alimentatii cu un aport suficient de hidrati de carbon.[6]
Diabetul renal fosfaturic
Excretia renala a fosfatilor se face prin filtrare glomerulara si reabsorbtie tubulara. Nu s-a dovedit existenta unor mecanisme de secretie tubulara a fosfatilor. La niveluri fiziologice ale fosfatemiei, ultrafiltrarea fosfatilor anorganici este completa. Deoarece s-a dovedit ca excretia fractionata este de 10-15% din cantitatea filtrata, rezulta ca 85-90% din fosforul anorganic se reabsoarbe tubular (60-70% in tubul proximal, 5-10% in nefronul mediu, 10-20% in tubul distal).
Cel mai important reglator al excretiei fosfatilor esteparathormonul (PTH). Cresterea nivelului plasmatic al PTH determina cresterea fosfaturiei fara cresterea filtrarii glomerulare. Hormonul are efect inhibitor asupra reabsorbtiei tubulare renale a fosfatilor atat proximal cat si distal.
Spre deosebire de PTH, vitamina D are rol antifosfatemic.
Transportul tubular al fosfatilor poate fi afectat prin:
Absenta hormonului efector
Nereceptivitatea
tubilor
Defecte structurale ale PTH
Absenta proteinei transportoare a fosfatilor
Absenta sau defectele enzimelor intracelulare necesare tranportului fosfatilor
Alterarea proceselor producatoare de energie la nivelul epiteliului tubular
Afectiunile care afecteaza transportul tubular al fosfatilor pot determina:
a. Pierdere urinara de fosfati cu hipofosfatemie si rahitism (la copil) sau osteomalacie (la adult);
b. Scaderea excretiei urinare de fosfati cu hiperfosfatemie si anomalii scheletice
Alterarea transportului tubular al fosfatilor se intalneste in:
Rahitismul vitaminoD-rezistent
Rahitismul vitaminoD-depenndent
Rahitismul vitaminoD- deficient (carential)
Rahitismulul hipofosfatemic al adultului
Rahitismulul oncogen
Hipoparatiroidism
Pseudohipoparatiroidism
Sindromul Fanconi si alte tubulopatii7
Rahitismul vitaminoD-rezistent (RVDR)
Definitie
RVDR este o maladie familiala caracterizata fiziopatologic prin defect de reabsorbtie tubulara si intestinala a fosfatilor, clinic prin rahitism sau osteomalacie, biologic prin hipofosfatemie si hipofosfaturie si terapeutic prin rezistenta la doze mari de vitamina D.
Patogenie
Anomalia fiziopatologica majora consta in scaderea reabsorbtiei tubulare renale proximale a fosfatilor si scaderea absorbtiei intestinale a calciului si fosforului. Nivelurile PTH si ale vitaminei D sunt normale.
Au fost propuse mai multe ipoteze pentru explicarea acestor tulburari, insa cele mai multe studii pledeaza pentru prezenta unei tubulopatii caracterizate printr-un defect primar interesand tranportul transmembranar al fosfatilor cu hiperfosfaturie consecutiva. Date mai noi au dovedit ca exista o tulburare a transportului intestinal al fosfatilor si sugereaza existenta unui defect de ordin general interesand transporturile prin membranele celulare.
Desi excesul de PTH joaca un rol minor in hiperfosfaturie, prezenta PTH este necesara pentru exprimarea defectului tubular, deoarece supresia PTH amelioreaza hipofosfaturia.[7, 8]
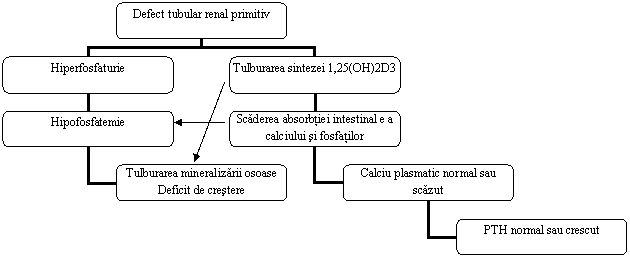

Figura 8.1 PatogeNefropatii interstitiale acute rahitismului vitaminoD-rezistent (adaptat dupa Ursea N (sub redactia),Tratat de Nefrologie, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006)
Anatomie patologica
Leziunile osoase constau in:
Proliferarea celulelor cartilajelor de crestere;
Largirea osteoidului;
Absenta mineralizarii, mai ales in jurul osteocitelor;
Reabsorbtie osteoclastica;
Deformari ale osului compact la nivelul metafizelor oaselor lungi cu tulburari ale structurii lamelare;
Pseudo-fracturi sau fracturi ale oaselor lungi; [7, 8]
Tablou clinic
Copii prezinta doua grupe principale de manifestari clinice: incetinirea cresterii si deformarile osoase.
Lipsesc miopatia, tetaNefropatii interstitiale acute si convulsiile intalnite in rahitismul vitamino-sensibil (carential).
Nanismul este disarmonic, micromelic (capul, gatul si trunchiul sunt de talie normala, membrele inferioare sunt scurte, deformate).
Deformarile osoase sunt mai accentuate la membrele inferioare si coloana vertebrala. Pot aparea: genum varum, genum valgum bilateral, sau genum varum de o parte si genum valgum de cealalta parte. Apar frecvent si tumefieri epifizare cu aspect de bratari la nivelul articulatiilor radio-cubito-carpiene si tibio-tarsiene, precum si tumefieri condro-costale cu aspect de matanii. Sunt prezente deformari ale bazinului: lordoza exagerata sau cifoscolioza.
Faciesul este particular cu fruntea bombata, radacina nasului larga, aspect antimongoloid al fantelor palpebrale.[7]
Evolutie
Activa pana la adolescenta, maladia intra apoi in faza de latenta. Bolnavul ramane un nanic cu deformari osoase multiple. Uneori se constata reprize evolutive in timpul sarcinii, alaptarii sau dupa 40 de ani.
Paraclinic
Hipofosfatemie (mai ales in prezenta unui istoric familial de RVDR)
Hiperfosfaturie
Calcemia normala sau usor crescuta
Fosfataza alcalina serica moderat sau net crescuta
PTH normal sau usor crescut
Nivelul vitaminei D normal sau usor scazut.[8]
Radiologic
Leziuni ale metafizelor identice cu cele din rahitismul carential : marirea metafizelor, aspect de cupola, margini neregulate ale platoului metafizar, zona anormal de larga ce separa nucleul metafizar de epifiza.
Anomalii ale texturii osoase, mai evocatoare pentru aceasta forma de rahitism. Ele constau in aspect mai lax, spatiat al trabeculelor. Afecteaza mai ales bazinul, extremitatile superioare si inferioare ale femurului, tibiei si humerusului, oasele carpiene.
Diagnostic pozitiv
Prezenta hipofosfatemiei si al istoricului familial de RVDR la un copil orienteaza diagnosticul. Ulterior trebuie stabilita vitamino-rezistenta (rahitismul carential raspunde la administrarea 2000 UI vitamina D3, zilnic, prin normalizarea fosfatemiei dupa 7-12 zile)
Odata stabilita vitamino-rezistenta trebuie eliminate mai multe afectiuni: malabsorbtie, tubulopatie mixta, acidoza tubulara distala, boala cronica de rinichi.[7]
Diagnostic diferential
Rahitismul vitaminoD-depenndent
Rahitismul vitaminoD- deficient (carential)
Rahitismul hipofosfatemic al adultului
Rahitismul oncogen
Sindromul Fanconi
Osteopatia hipofosfatemica
Hipofosfatemia din tubulopatiile mixte [7]
Tratament
Vitamina D in doze mari 20.000-40.000 UI/zi pana la 300.000-400.000 UI/zi. Rezultatele au fost modeste.
Suplimentarea orala cu fosfati la fiecare 4 ore, in doze egale. Acestia determina insa
hipocalcemie si hipoparatiroidism secundar si de aceea trebuie asociati cu vitamina D.
Monitorizarea terapiei se face prin determinari ale calcemiei, fosfatemiei, caciuriei, fosfaturiei, fosfatazei alcaline, la 15 zile in prima luna, lunar in primul trimestru si o data la 2-3 luni in primul an de tratament.[7]
Rahitismul vitaminoD-dependent
Patogenie
Afectiunea se datoreaza unei deficiente enzimatice ereditare in 1α-hidroxilaza, care converteste 25(OH)D3 in 1,25(OH)2D3. Este cea mai intalnita afectiune ereditara care evolueaza cu hipofosfatemie. Reprezinta 80% din cazurile de sindroame ereditare familiale cu pierdere urinara de fosfati. Este determinata de o mutatie a genei PHEX, care este exprimata in osteoblaste si in tumori tisulare ce au hipofosfaturia ca manifestare paraneoplazica. [9]
Tablou clinic
Debutul bolii are loc in jurul varstei de 3-6 luni, cu manifestari tetanice, convulsii, hipotonie musculara. Apar ulterior deformari osoase de tip rahitic. Cresterea staturo-ponderala si dezvoltarea psiho-motorie sunt intarziate.[7]
Paraclinic
Hipocalcemie severa
Hipofosfatemie
Hiperfosfaturie
PTH seric crescut
AMPc seric crescut
Hiperaminoacidurie [8]
Tratament
Se administreaza doze mari de vitamina D3 (100.000-200.000 UI) sau doze mici de 1,25(OH)2D3 (1 microg/zi).[7]
Rahitismul vitaminoD-eficient (osteopatia hipofosfatemica fara rahitism)
Evolueaza cu deficit statural moderat, dar fara rahitism, precum si cu hipofosfatemie fara hiperfosfaturie. Transportul tubular al fosfatilor este normal.
Rahitismul hipofosfatemic al adultului
Debutul are loc dupa varsta de 15 ani, cu astenie si hipotonie musculara, dureri si deformari osoase. Apar cifoza si scolioza, iar biologic hipofosfatemie severa, calcemie normala sau usor scazuta, fosfaturie crescuta, calciurie normala sau redusa. Valorile PTH si 25(OH)D3 sunt normale. PatogeNefropatii interstitiale acute este necunoscuta, iar tratamentul cu fosfati si doze mari de vitamina D nu produce vindecarea.[7]
Rahitismul oncogen
Se asociaza cu tumori mezenchimale benigne: tumori osoase, displazii fibroase, neurofibromatoza, hemangiom cavernos, tumori cu celule gigante. Se caracterizeaza prin rahitism, hipofosfatemie, calcemie normala, PTH seric normal, reabsorbtie tubulara a fosfatilor scazuta. In patogenie se presupune ca ar interveni substante secretate de tumora. Dupa excizia tumorii fosfatemia se normalizeaza [8]
Pseudohipoparatiroidismul (Osteodistrofia ereditara Albright)
Definitie
Este un sindrom familial caracterizat clinic prin anomalii scheletice, biologic prin hipocalcemie si hiperfosfatemie in ciuda valorilor crescute de PTH si patogenic prin nereceptivitatea organelor tinta la actiunile PTH.
Patogenie
Organele tinta nu sunt sensibile la actiunea PTH, o cauza probabila fiind deficienta adenilatciclazei PTH-sensibile.
Tablou clinic
Pacientul prezinta:
Statura scunda
Facies rotund si gat scurt
Obezitate
Metacarpienele si metatarsienele 4 si 5 scurte
Calcificari subcutanate
Tetanie hipocalcemica
Strabism, cataracta
Retard mental [7]
Paraclinic
Hipocalcemie ( datorata scaderii absorbtiei intestinale de calciu si scaderii raspunsului osos la actiunea PTH)
Hiperfosfatemie (secundara lipsei de raspuns a tubului proximal la actiunea PTH)
Sindromul Toni-Debr-Fanconi
Definitie
Reprezinta o disfunctie generalizata a tubului proximal, fara a fi asociata cu afectare glomerulara primara, caracterizata prin pierdere urinara crescuta de fosfati, glucoza AA si bicarbonati.[10]
Sindromul Fanconi poate fi ereditar ( idiopatic-afectare primara a tubului proximal, sau secundar acumularii unor substante toxice la nivel renal in cadrul altor afectiuni metabolice ereditare) sau dobandit ( asociat cu sindrom nefrotic, paraproteinemii sau diverse toxice).[10]
Clasificare etiologica
Sindrom Fanconi ereditar:
A. Idiopatic
B. Secundar ( cistinoza, tirozinemia, galactozemia, Boala depozitelor de glicogen, Boala Wilson)
Sindom Fanconi dobandit:
A. Mielom multiplu
B. Sindrom nefrotic
C. Nefropatii tubulointerstitiale cronice
D. Transplant renal
E. Factori exogeni (Ca, Hg, Pl, cisplatin, aminoglicozide) [11]
Tablou clinic
Aminoaciduria
Din punct de vedere clinic, excretia crescuta de aminoacizi (AA) nu duce la deficite grave si nu necesita suplimentarea aportului oral al acestora.[10]
Fosfaturia si boala osoasa
Pierderea de fosfati reprezinta cea mai importanta manifestare a sindromului. Apar deformari osoase mai ales la membrele inferioare, iar la adulti dureri osoase si fracturi spontane.[10]
Acidoza tubulara renala
Este o acidoza hipercloremica, care necesita doze crescute de bicarbonat pentru corectie.[10]
Glicozuria
Nivelul glicemiei ramane normal, in timp ce pierderile urinare sunt de 0.5-10 g/zi. Glicozuria masiva si hipoglicemia secundara poate fi intalnita in diabetul nefrogen tip B. [12]
Pierderea renala de Na si K
Pot determina hipotensiune, hiponatremie, alcaloza metabolica. Se corecteaza prin administrarea de NaCl si saruri de K.[10]
Proteinuria
De obicei este scazuta sau moderata. Pentru a demonstra originea tubulara a acesteia poate fi dozata excretia de β2-microglobulina.[10]
Hipercalciuriile idiopatice
Reprezinta un grup de afectiuni caracterizat prin excretia urinara zilnica de Ca de peste 300 mg/zi la barbat si de peste 250 mg/zi la femeie, fara o alta cauza de hipercalciurie (hipercalcemie, sarcoidoza, acidoza tubulara, neoplazii, boala Paget etc). Acesti pacienti nu au aport crescut de vitamina D sau calciu, dar au o absorbtie intestinala crescuta de calciu.
Excretia zilnica este, in medie, dubla fata de normal. Acest lucru ar putea fi determinat, fie de marirea cantitatii de calciu filtrate, fie de scaderea reabsorbtiei calciului filtrat, fie prin asocierea ambelor mecanisme.
Pe baza cercetarilor efectuate, Coe si Bushinski, au concluzionat ca hipercalciuria idiopatica poate fi consecinta fie a unei cresteri a absorbtiei intestinale de calciu secundare unei anomalii de sinteza a 1,25(OH)2D3, fie a unei diminuari primitive a reabsorbtiei tubulare renale de calciu, autorii fiind in favoarea primei ipoteze.
Importanta practica a acestei afectiuni rezida in faptul ca este mult mai frecventa la pacientii cu litiaza calcica, reprezentand primul factor de risc metabolic la acesti pacienti.[10]
Tulburarile transportului tubular al aminoacizilor
Aminoacizii sunt in intregime filtrabili si in cea mai mare parte se reabsorb la nivelul tubului proximal. Reabsorbtia se face prin mecanisme de transport activ, a caror caracteristica este prezenta unui sistem tranportor specific (carrier) situat la nivelul membranei celulare. Exista doua sisteme de transport pentru aminoacizi din lumen la celula tubulara:
i. pentru un singur AA cu capacitate joasa si specificitate inalta
ii. specific de grup, cu capacitate mare si specificitate redusa
Clasificarea hiperaminoaciduriilor (dupa mecanismul de producere):
A. Prerenale:
a. Hiperaminoaciduria
de saturare (overflow)
asociata cu hiperaminoacidemie, intalnita
b. Hiperaminoaciduria
prin competitie asociata
cu hiperaminoacidemie, intalnita
c. Hiperaminoaciduria
fara prag care nu se
asociaza cu hiperaminoacidemie, intalnita
B. Renale:
a. Prin deficit al sistemelor de transport al unui singur AA (hipercistinuria, hiperglicinuria, malabsorbtia metioninei);
b. Prin deficit al sistemelor de transport specific de grup (cistinuria, boala Hartnup, iminoglicinuria);
c. Prin deficit global al transportului tubular (hiperaminoacidurii generalizate).
De obicei sunt depistate la nou-nascuti, cel mai frecvent intalnite fiind cistinuria, cistidinuria, boala Hartnup, iminoglicinuria. Cea mai importanta din punct de vedere clinic este hipercistinuria, celelalte fiind rareori simptomatice.[13]
Cistinuria
Se caracterizeaza printr-un defect mostenit de reabsorbtie tubulara a cistinei si a aminoacizilor dibazici la nivelul tubului proximal.
Au fost identificate trei tipuri de cistinurie:
Tipul I (forma recesiva completa) cauzat de mutatii ale genei SLC3A1.
Tipul II si III (forme recesive incomplete), legate de un locus de pe cromozomul 19q13.1, gena nefiind inca identificata.
Nefrolitiaza apare cand excretia de cistina depaseste capacitatea ei de a se solubiliza la un pH urinar scazut in tubul distal.[14]
Acidozele tubulare renale
Definitie
Acidozele tubulare renale (ATR) sunt defecte tubulare renale care au in comun incapacitatea tubilor de a acidifia normal urina. ATR sunt sindroame caracterizate prin acidoza metabolica hipercloremica cu pH urinar anormal de mare fata de gradul acidemiei. Acidoza tubulara renala este o conditie caracterizata prin deficit de excretie a ionilor de H+, de reabsorbtie a bicarbonatilor, sau ambele, in prezenta unei functii glomerulare normale.
Clasificare
In functie de sediul tubular si mecanismele patogenice implicate, se distig doua grupe mari de ATR:
A. ATR distala de tip I, determinata de incapacitatea tubului distal de a stabili un gradient adecvat de pH intre plasma si lichidul tubular.
B. ATR proximala de tip II, determinata de deficitul de absorbtie al bicarbonatilor la nivelul tubului proximal
C. ATR de tip III sau distala. ATR de tip I se asociaza cu o pierdere mai mult sau mai putin importanta de bicarbonati, care poate fi tranzitorie si se poate ameliora cu varsta.
D. ATR de tip IV, caracterizata prin prag scazut al bicarbonatilor, deficit de mineralocorticoizi si hiperkaliemie.
E. ATR tip I-II hibrid
Dupa criteriul etiologic se disting ATR primitive si secundare (boli ale metabolismului calciului, boli autoimune si disglobulinemii, cauze toxic-medicamentoase).[15, 16]
Tubulopatii ale ansei Henle
Sindromul Bartter (alcaloza hipokaliemica familiala) si sindromul Gitelman
Definitie
Sunt afectiuni transmise autozomal recesiv, caracterizate prin hipokaliemie ereditara, alcaloza metabolica, depletie considerabila a volumului intravascular, datorita unor mutatii heterogene care afecteaza co-transportorul Na+/K+/Cl- sensibil la bumetanid sau canalul de K+ ATP reglat sau canalul de Cl- voltaj dependent CLC-Kb (sindrom Bartter), sau co-transportorul Na+/Cl- sensibil la tiazidice (sindrom Gitelman).
Mai multi autori au propus clasificarea acestor bolnavi in mai multe categorii, in functie de excretia urinara de calciu:
bolnavi cu hipocalciurie, care sunt considerati a avea sindrom Gitelman
bolnavi cu hipercalciurie considerati a avea sindrom Bartter.[17]
Anatomie patologica
Biopsia renala a evidentiat constant o hipertrofie si hiperplazie a aparatului juxtaglomerular (AJG). Frecvent se intalnesc si vacuolizarea nefrocitelor proximale, dilatari ale tubilor distali cu atrofia epiteliului.
Clinic
Simptomul revelator este poliuria, responsabila de deshidratare acuta in copilarie. Hipotrofia staturo-ponderala este notabila si intarzierea cresterii se accentueaza, ajungand repede la nanism. Deasemenea mai pot aparea crize comitiale, tetanie, slabiciune musculara, parestezii, dureri articulare, condrocalcinoza sau aparitia unei nefrocalcinoze cu boala cronica de rinichi.
Paraclinic
Incapacitatea de a concentra urina;
Hipokaliemie considerabila cu hiperkaliurie permanenta;
Alcaloza metabolica cu hipocloremie;
Hiperaldosteronism cu TA normala sau scazuta;
Excretie urinara crescuta de prostaglandine;
Diagnostic diferential
Sindromul pseudo-Bartter, la adulti dupa varsaturi, diuretice, laxative
Hiperaldosteronismul primar ( care este insotit de HTA)
Diareea familiala clorurica
Sindromul Fanconi (afectare tubulara complexa)
Tratament
Actual, nu exista un agent farmacologic lipsit de riscuri in tratamentul sindromului Bartter.
Pentru corectarea deficitului de K+ se administreaza saruri de potasiu oral. [17, 18, 19]
Hipomagnezemiile ereditare
Locul major de reabsorbtie pasiva al Mg este ramura larga ascendenta Henle unde 50-72% din Mg filtrat este reabsorbit.
Boli ereditare cu hipomagnezemie secundara
In aceasta categorie intra bolile tubulare ereditare cu alcaloza hipokaliemica la care poate fi prezenta si hipomagnezemia.
Hipomagnezemiile ereditare primitive
In patru boli ereditare distincte hipomagnezemia este anomalia dominanta. Este vorba de o afectiune cu defect de absorbtie intestinala a Mg si trei afectiuni cu anomalii de reabsorbtie renala a Mg²+.
A. Hipomagnezemia digestiva primitiva cu hipocalcemie secundara (HHS)
Se caracterizeaza prin magnezemie foarte scazuta cu excretie normala de Mg²+ si hipocalcemie. Este o afectiune letala in absenta tratamentului.
B. Hipomagnezemie, hipercalciurie si nefrocalcinoza (HHN)
Este o afectiune autozomal recesiva, recunoscuta din copilarie, caracterizata prin hipomagnezemie cu hipocalcemie, hipercalciurie complicata adesea cu nefrocalcinoza si boala cronica de rinichi. Mai pot aparea infectii, anomalii oculare, hiperuricemie, acidoza tubulara incompleta.
C. Hipomagnezemia cu hipocalciurie
Afectiunea este asemanatoare cu sindromul Gitelman, dar lipsesc hipokaliemia si alcaloza metabolica
D. Hipomagnezemie cu normocalciurie
Este vorba de un singur caz raportat de defect de reabsorbtie a Mg²+, fara o anomalie de metabolism a calciului.[21]
Tubulopatiile cronice distale
Diabetul insipid nefrogen
Definitie
Reprezinta nefropatie tubulara cu afectarea reabsorbtiei apei.
Clasificare
A. Diabet insipid nefrogen ereditar (DINE)
B. Diabet insipid nefrogen dobandit (DIND)
DINE este o afectiune ereditara cu transmitere recesiva legata de sex, in timp ce DIND este un sindrom ce apare in anumite circumstante: tulburari electrolitice (hipokaliemie, hipercalciurie), administrare de medicamente, amiloidoza, sindrom Sjogren, polichistoza renala, pielonefrita cronica. Din acest motiv adevaratul diabet insipid nefrogen trebuie considerat cel ereditar, forma dobandita fiind mai mult un sindrom poliuro-polidipsic cu etiopatogenie diferita.[22, 23]
Diabetul insipid nefrogen ereditar
Definitie
Tubulopatie primitiva cronica ereditara caracterizata prin lipsa receptivitatii tubilor distali la actiunea ADH endogen si exogen, care se manifesta clinic prin: poliurie, polidipsie, subizostenurie, deshidratare hipertona.
Nivelul de ADH plasmatic este mai crescut decat la subiectii normali.
Patogenie
Hormonul antidiuretic (arginin-vasopresina) este secretat la nivelul nucleilor supraoptici si paraventriculari hipotalamici si eliberat la nivelul hipofizei posterioare pe cale axonala, ca raspuns la cresterea osmolaritatii plasmatice sau hipovolemie.
La nivel extrarenal, regleaza TA impreuna cu sistemul RAA si sistemul nervos simpatic, fiind un puternic vasoconstrictor prin actiunea lui la nivelul receptorilor V1.
La nivel renal, actioneaza pe receptorii V2 determinand cresterea permeabilitatii pentru apa a tubilor distali si colectori.
La pacientii cu DINE, s-a postulat ipoteza ca defectul primar este absenta raspunsului tubului distal si colector la actiunea ADH, secundar deficitului de AMPc, deoarece nici nivelurile plasmatice crescute de ADH structural normal, nici administrarea de ADH exogen nu provoaca o crestere a osmolaritatii urinare .
Tablou clinic
Primele simptome apar in primele saptamani sau luni de viata, sau dupa trecerea la alimentatia artificiala, care, prin sarcina osmotica mai mare, agraveaza poliuria si deshidratarea.
Poliuria este primul semn care apare, insa este destul de greu de depistat.
Curand apar simptomele deshidratarii cronice: febra de cauza neexplicata, anorexie, varsaturi, constipatie, intarzierea cresterii. In 50-75% din cazuri, la nou-nascuti, nu exista polidipsie.
In timp pot aparea trei grupe de consecinte clinice: episoade de deshidratare severa, deficit staturo-ponderal si retardul mental.
Paraclinic
Hipernatremia peste 150 mEq/l,
Hipercloremia peste 120 mEq/l
Densitatea urinara 1001-1008 mmol/kg
Osmolaritatea plasmatica 50-100 mosm/kg
Hiperuricemie (in unele cazuri)
Diagnostic pozitiv
Diureza crescuta
Densitate urinara sub 1008
Osmolaritate urinara 15-160 mosm/kg
Lipsa capacitatii de concentrare a urinii la testul setei
Lipsa capacitatii de concentrare a urinii la proba cu solutie salina
Diagnosticul diferential
Se face in primul rand cu DIND, avand in vedere istoricul familial.
Tratament
Aport hidric corespunzator la intervale regulate, chiar si noaptea.
Dieta adecvata, in primele luni de viata cu lapte uman.
Tratamentul cu hidroclorotiazida 1-2 mg/kgcorp/zi este eficient inca de la administrarea primei doze. La fel de active sunt furosemidul si acidul etacrinic 2-3 mg/kgcorp/zi. Hiperuricemia se trateaza cu allopurinol.
4. Indometacina, inhibitor al PG, are efect antidiuretic in DINE.[24, 25, 26, 27]
Diabetul insipid nefrogen dobandit
Conditiile clinice in care poate aparea sunt:
1. Hipokaliemie, nefropatie hipokeliemica
2.Hipercalcemie, nefropatie hipercalcemica
3. Nefropatii interstitiale
4.Nefropatii malformative (polichistoza renala, boala medulara chistica)
5. Uropatii obstructive
6. Disglobulinemii si boli autoimune (mielom multiplu, poliartrita reumatioda, amiloidoza, ciroza hepatica)
7. Malnutritie
8. Cauze toxic-medicamentoase ( Amfotericina B, Pb, Li, Etanol, Fenitoina)
In toate aceste cazuri, ancheta familiala este negativa, defectul de concentrare al urinii este reversibil, odata cu tratarea bolii de baza. [28]
Pseudohipoaldosteronismul familial (sindromul Cheek-Perry)
Definitie
Este o tubulopatie ereditara rara, de etiologie necunoscuta, caracterizata prin non-receptivitatea tubilor distali la aldosteron, pierderi urinare excesive de Na si Cl, hiponatremie, hiperkaliemie.
Clinic
Debutul afectiunii are loc in primele zile de viata cu anorexie si ocazional varsaturi. Copii au curba ponderala stationara, hipotonie musculara si panicul adipos redus. Apar si leziuni cutanate, eritem scrotal. Bolnavii au diureza normala sau poliurie moderata. Examenul obiectiv evidentiaza copii apatici, somnolenti, cu semne de deshidratare.
Paraclinic
Diagnostic pozitiv
Se formuleaza pe baza datelor clinice si paraclinice de mai sus.
Diagnosticul diferential
Trebuie discutat in cazul:
Tratament
Suplimentarea orala de NaCl (3-6 g/zi) amelioreaza simptomatologia.
3. Pseudohipoaldosteronismul familial (sindrom Liddle)
Este o tubulopatie cronica ereditara cu transmitere autozomal dominanta caracterizata prin HTA, hipokaliemie, alcaloza metabolica, defect de concentrare a urinii, niveluri serice scazute de aldosteron seric si urinar.
Diagnosticul diferential se face cu hiperaldosteronismul primar si sindromul Bartter, care au aldosteron seric crescut.
Tratamentul consta in restrictie de Na, administrarea orala de K si triamteren.[29]
Bibliografie
1. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3311, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
2. Ciocalteu A (sub redactia), Tratat de Nefrologie, 333, Editura National, Bucuresti, 2006
3. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3314-3315, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
4. Loeffler J, Sakallioglu O, et al, Familial renal glucosuria: SLC5A2 mutation and evidence of salt-wasting. Kidney Int 69:852-855, 2006
5. Cooper C, Heilig CW, Identification of a novel form of renal glucosuria with overexcretion of arginine, carnosine and taurine. Am J Kidney Disease, 1038-1043, 2001
6. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3322-3325, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
7. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3325-3335, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
8. Brenner & Rector`s - The Kidney - 8th edition, 1400-1405, SAUNDERS ELSEVIER ,2008
9. Beck J, Soumounou Y, Martel J et al, Pex/PEX tissue distribution and evidence of a deletion in the 3' region of the Pex gene in X-linked hypophosphatemic mice,Invest 99:1200-1209, 1997
10. Brenner & Rector`s - The Kidney - 8th edition, 1406-1418, SAUNDERS ELSEVIER ,2008
11. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3368-3374, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
12. Sanjad SA, Kaddoura RE, Nayer HM, et al, Fanconi's syndrome and glicogenosis associated with phosphorilase B kinase deficiency, 147:957-959, 1993
13. Wilcken B, Smith A, Brown DA, Urine screening for aminoacidopathies: is it beneficial? Results of along-term follow-up f cases detected by screening one milion babies. J Pediatr 97:492-497, 1980
14. Ursea N, Actualitati in Nefrologie, Editura Fundatiei "RomaNefropatii interstitiale acute de Maine", 2000
15. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3374-3390, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
16. Brenner & Rector`s - The Kidney - 8th edition, 1408-1409, SAUNDERS ELSEVIER ,2008
17. Brenner & Rector`s - The Kidney - 8th edition, 1409-1410, SAUNDERS ELSEVIER ,2008
18.Bettinelli A, Bianchetti M, Girardin E, Caringella A, Cecconi M, Applani AC, Pavanello L, Gastaldi R, Lama G, Use of calcium excretion values to distinguish two form of primary renal tubular Hypokaliemic alkalosis: Bartter and Gitelman Syndrome, J Pediatr, 1992, 120, 38-43
19. Clive MD. Bartter syndrome: the unsolved puzzle. AM J Kidney Dis, 1995, 25, 813-823
20. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3400-3404, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
21. Botero Veley M, Curtis JJ, Warnock DG. Liddle's syndrome revised. A disorder of sodium reabsorbtion in the distal tubule. N Engl J Med 1994, 330, 178-181
22. Ursea N (sub redactia), Tratat de Nefrologie, Volumul 3, 3406-3411, Editura Fundatiei Romane a Rinichiului, Bucuresti, 2006
23. Brenner & Rector`s - The Kidney - 8th edition, 1417-1419, SAUNDERS ELSEVIER ,2008
24..Knoers N, Monneens LAH, A variant of nephrogenic diabettes insipidus: V2 receptor abnormality rescricted to the kidney. Eur J Pediatr, 1991, 150, 370-373
25. Knoers N, Monneens LAH, Nephrogenes diabetes insipidus:clinical symptoms, pathogenesis and treatment, Pediatr Nephrol, 1992, 6, 476-482
26..Langlez JM; Balfe JW, Selander T, Raz PN; Clarke JTR, Autosomal recessive inheritence of nephogenic diabetes insipidus, Am J Hum Genet, 1991, 38, 90-94
27..McKusickVA, Diabetes insipidus renal type I, Mendelian inheritance in man, 9-th
ed.
28. Brenner & Rector`s - The Kidney - 8th edition, 1420-1422, SAUNDERS ELSEVIER ,2008
29. Brenner & Rector`s - The Kidney - 8th edition, 1412-1413, SAUNDERS ELSEVIER ,2008
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3177
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved