| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
POLIGLOBULIA PRIMITIVA (POLICITEMIA VERA) (PV)
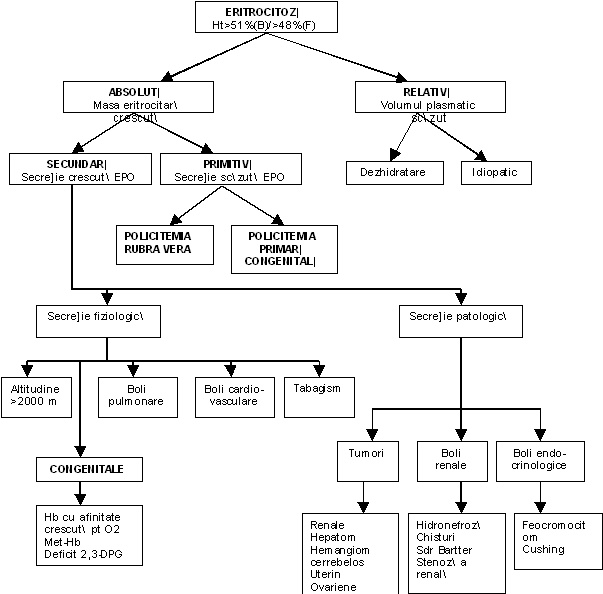
Termenul de eritocitoza define te situatiile in care valoarea hemoglobinei depaseste limita superioara a normalului sau in care valoarea hematocritului depaseste 51% la barbati si 48% la femei. Poliglobulia sau policitemia sunt adesea utilizate ca termeni sinonimi cu eritrocitoza dar, mai corect, primii termeni se refera la o crestere a mai mult de o linie celulara hematopoietica. Eritrocitoza se subimparte in eritocitoza absoluta (insotita de cresterea masei eritrocitare totale) si in eritocitoza aparenta (neinsotita de cresterea masei eritocitare totale). La nivelul marii, masa eritrocitara normala este de 23-29 ml/kg greutate corporala, pentru femei si de 26-32 ml/kg greutate corporala, la barbati.
In cazul in care eritocitoza aparenta se asociaza cu o scadere a volumului plasmatic este denumita ca eritrocitoza relativa. Eritocitoza aparenta nu se insoteste intotdeauna de o scadere a volumului plasmatic si poate rezulta din cresterea raportului intre masa eritrocitara si volumul plasmatic.
In cazul in care eritocitoza absoluta reprezinta o boala hematologica clonala asociata cu cresteri si al altor linii mieloide, este denumita ca eritrocitoza (poliglobulie sau policitemie) primitiva. Eritrocitoza absoluta neclonala datorata supraproductiei fiziologice sau patologice de eritropoietina, este denumita eritrocitoza secundara.
Eritrocitoza primitiva reprezinta o proliferare autonoma a celulelor progenitoare ale liniei eritrocitare, datorata unui defect celular intrinsec sau , mai rar, unei alterari a controlului eritropoiezei. Eritrocitoza primitiva cuprinde doua forme: policitemia vera si eritrocitoza primitiva pura (eritremia).
Eritrocitoza primitiva pura reprezinta un sindrom heterogen caracterizat prin cresterea izolata a masei eritrocitare fara stigmatele policitemiei vera si fara cauze aparente de eritrocitoza secundara. Au fost descrise cazuri familiale si cazuri sporadice. Nivelurile serice de eritropoietina sunt normale sau scazute. Unele cazuri evolueaza spre poliglobulie primitiva.
Policitemia vera reprezinta un sindrom mieloproliferativ de etiologie necunoscuta, survenit prin implicarea celulei stem pluripotente. Se caracterizeaza prin hiperproductie predominanta sau exclusiva de globule rosii antrenand o poliglobulie. Proliferarea are caracter monoclonal. Expansiunea precursorilor eritrocitari este o proprietate intrinseca a celulelor si este independenta de sau hipersensibila la controlul exercitat de factorul de crestere, eritropoietina (EPO). Mecanismul de producere ramane necunoscut. La unii pacienti cu eritrocitoza familiala au fost implicate mutatii somatice la nivelul genei receptorului pentru EPO.
Caracterul monoclonal al bolii explica potentialul sau de transformare in leucemie acuta sau de a dezvolta o fibroza medulara datorita secretiei anormale de factori de crestere ai fibroblastilor de catre megakariocitele clonale. Asocierea policitemiei cu o crestere a riscului trombotic este predominant corelata cu cresterea vascozitatii sanguine datorata cresterii hematocritului. Ulterior, interactiunea peretelui vascular cu numarul crescut de leucocite sI trombocite clonale poate antrena o adezivitate leucocitara crescuta cu activare trombocitara conducand la diateza trombotica din cadrul bolii.
Eritrocitoza primitiva si cea secundara pot apare ca boli dobandite dar si ca forme congenitale.
Policitemia vera este o boala rara, mai ales la rasa alba, avand o prevalenta de 0,2-5 cazuri la o populatie de 100.000 persoane. Icidenta este foarte scazuta in Africa si Asia (2 cazuri la 1 milion de persoane in Japonia). Poate aparea si ca forma familiala. Boala poate surveni la orice varsta, dar este rar semnalata la copii. Mediana varstei la debut este in jurul celei de 60 ani. Exista o discreta predominanta masculina (sex ratio= 1,2).
Tabelul I
Debutul bolii este lent, insidios, adesea descoperirea fiind intamplatoare. Semnele si simptomele fiind nespecifice, diagnosticul este adesea tardiv in raport cu momentul debutului. Debutul se poate produce si direct, brutal, prin complicatii (tromboze, hemoragii).
Semnele si simptomele se datoreaza hipervascozitatii sanguine (datorate poliglobuliei) si/sau complicatiilor din domeniul hemostazei.
Ø tegumente si mucoase :
eritroza faciala cu nuanta rosie purpurie, predominanta la nivelul buzelor, pometilor, nas, urechi, gat, eritroza mucoaselor, predominant conjunctivale ;
echimoze ;
tegumente uscate, acnee ;
prurit intens, rebel, adesea declansat dupa bai calde - apare la peste 45 % din pacienti - manifestare rar semnalata in formele secundare .
Ø sistem nervos :
cefalee, vertije, lipotimii ;
acufene, scotoame, amauroza tranzitorie ;
paralizii, pareze ;
accidente vasculare cerebrale cu mono- sau hemiplegii, afazie ;
tulburari de comportament.
Ø splenomegalie (75%) cu sau fara hepatomegalie (40%)
Ø manifestari cardio-vasculare - HTA moderata, tromboze arterio-venoase
Ø manifestari digestive - ulcer gastric sau duodenal, hemoragii, infarct mezenteric, sdr Budd-Chiari.
Trebuie remarcate cateva semne negative :
absenta semnelor de insuficienta respiratorie ;
absenta unei hepatomegalii tumorale ;
lojele renale sunt libere ;
absenta semnelor neurologice de sindrom tumoral de fosa posterioara.
Ø Hemograma
Hb > 18g/dl, Ht* > 55%, GR > 6.000.000/mm3, normocromie, normocitoza ;
leucocitoza inconstanta cu GA > 12.000/mm3, cu fosfataza alcalina leucocitara (FAL) crescuta, in formula predomina neutrofilele ;
prezenta unei mielemii, cu cresterea eozinofilelor si bazofilelor, sugereaza un sindrom mieloproliferativ ;
hiperplachetoza cu Tr > 400.000/mm3, uneori pana la 1-3 x106/mm3, cu afectare functionala (scaderea adezivitatii si agregabilitatii) si alungirea timpului de sangerare.
Confirmarea poliglobuliei se face prin masurarea masei eritrocitare totale (volumul eritrocitar total). Metoda de referinta ramane masurarea volumului sanguin total cu hematii marcate cu Crom51 si determinarea volumelor globulare si plasmatice cu ajutorul valorilor microhematocritului. Valorile obtinute arata o crestere cu peste 20-25% fata de limita normala superioara. Astfel exprimat, se obtin valori de peste 36 ml/kg la barbati si peste 32 ml/kg la femei.
Ø Mielograma - arata o maduva de aspect normal, bogata in megacariocite. Nu este necesara pentru diagnostic.
Ø Biopsia medulara
Permite evaluarea celularitatii medulare, analiza cantitativa si calitativa a fiecarei linii mieloide si studiul retelei reticulinice.
In 90% din cazuri, biopsia medulara va confirma diagnosticul aratand o maduva bogata, hiperplazica (cu celularitate 60-100%), cu disparitia spatiilor grasoase, hiperplazia celor trei linii, cu predominanta liniilor eritroblastica si megakariocitara cu semne de dismegakariopoieza, hiperplazie granulocitara, accentuarea tramei de reticulina si absenta depozitelor de fier.
In poliglobulia secundara, maduva are aspect hipocelular, reactiv.
Examenul repetitiv al maduvei poate fi util pentru a surprinde precoce, evolutia spre mielofibroza.
Ø Biochimic
VSH = 0 sau mult scazut
cresterea concentratiei plasmatice a vitaminei B12 si a transcobalaminei
hiperuricemie
hemostaza perturbata cu alungirea timpului de sangerare si scaderea agregabilitatii plachetare.
pseudo-hiperpotasemie datorata eliberarii de potasiu din trombocite in cursul coagularii in vitro la pacientii cu trombocitoza.
Pentru eliminarea sau diferentierea de o poliglobulie secundara trebuie realizate cateva investigatii suplimentare :
gazele in sangele arterial, cu o presiune a oxigenului (PaO2) peste 65 mmHg, si o saturatie in oxigen (SaO2) peste 92%
determinarea afinitatii hemoglobinei pentru oxigen (P50 - presiunea oxigenului la care hemoglobina devine saturata 50 %) - la pacientii cu hemoglobinopatii cu afinitate crescuta pentru oxigen, P50 este scazut (mai putin de 20 mmHg fata de un normal de 27,5 mmHg). Este scazut si in carboxihemoglobina dar este normal in PV
dozarea fosfatazei alcaline leucocitare (FAL) - FAL este o enzima prezenta in granulatiile secundare ale neutrofilelor. Este crescuta in peste 70 % din cazurile de PV, permitand diferentierea de o LMC ;
echografie +/- scanner hepatic - pentru depistarea unui eventual hemangiom sau tumora hepatica ;
ecogafie renala, urografie - pentru depistarea unei eventuale tumori renale ;
scanner cerebral (eventual) - pentru evidentierea unei eventuale tumore cerebeloase ;
dozajul eritropoietinei serice si urinare - este crescuta la cei cu forme secundare si scazuta la cei cu forma primitiva
culturi celulare de progenitori eritroblastici (BFU-E) - la pacientii cu poliglobulie primitiva progenitorii cresc in cultura fara a necesita adaugarea de eritropoietina in mediul de cultura (proliferare autonoma).
Ø Citogenetica - examenul citogenetic al pacientilor cu PV evidentiaza anomalii cromozomiale clonale la peste 30 % din pacientii netratati si la peste 50 % din cei tratati. Anomaliile pot fi prezente la debut sau pot apare in evolutie. Anomaliile cele mai frecvent semnalate sunt deletiile bratului lung al crs 5(5q-), ale bratului lung al crs 20(20q-), trisomia 8, trisomia 9. Cromozomul Philadelphia nu este prezent.
Semnele si simptomele sunt multiple dar nepatognomonice astfel incat diagnosticul se face pe baza asocierii unor criterii stabilite de Polycytemia Vera Study Group (PVSG).
Tabelul III
Criterii de diagnostic PVSG |
|
Majore (A) |
|
A1 - volum globular total > 36 ml/kg la barbat si > 32 ml/kg la femeie A2 - saturatia arteriala in oxigen > 92% A3 - splenomegalie |
|
Minore (B) |
|
B1 - trombocitoza : Tr > 400.000/mm3 B2 - leucocitoza : GA > 12.000/mm3 in absenta infectiei sau febrei B3 - scorul FAL > 100, in absenta infectiei sau febrei B4 - vitamina B12 serica >900 pg/ml (n = 130-875 pg/ml) B5 capacitatea de legare vit B12 > 2200 pg/ml |
|
Diagnostic = A1 + A2 + A3 sau A1 + A2 + 2B |
A1 confirma existenta unei eritrocitoze absolute. A2 elimina contextul cel mai reprezentativ de aparitie a unei poliglobulii secundare. A3, B1-4 confirma un context mieloproliferativ.
Recent OMS a stabilit noi criterii de diagnostic pentru PRV:
Diagnosticul este stabilit prin prezenta criteriilor A1 si A2 impreuna cu oricare alt criteriu A sau cu doua din criteriile B.
Se face cu falsele poliglobulii si cu poliglobuliile secundare, eventual cu celelalte sindroame mieloproliferative (vezi mai jos).
In absenta tratamentului, decesul survine, in medie, dupa 18 luni de evolutie. Decesul survine, in majoritatea cazurilor, de cauza vasculara. Sub tratament evolutia se poate prelungi pana la 12-14 ani.
v Complicatii vasculare
tromboze si tromboembolism - survin la 15 - 60 % din cazuri si reprezinta principala cauza de deces (la 10 - 40 % dintre ei). Sunt favorizate de vascozitatea sanguina (exista o corelatie intre valoarea hematocritului si riscul trombotic), hiperleucocitoza si hiperplachetoza. Trombozele pot fi venoase sau arteriale. Mai frecvent survin : tromboza cerebrala, coronariana, pulmonara, mezenterica, tromboze arteriale ale membrelor, embolii pulmonare. Factorii de risc pentru tromboze sunt reprezentati de varsta peste 70 ani, antecedente trombotice, bolile vasculare, flebotomiile repetate. Astfel, la pacientii asimptomatici asociind factori de risc vascular si sistematic la cei peste 70 ani trebuie explorate vasele principale pentru a preveni accidentele tombotice.
hemoragii - survin la 20 - 35 % din cazuri si sunt cauza de deces la 10 - 25 % din pacienti. Sunt favorizate de trombopatie si de dilatatia vasculara antrenata de poliglobulie. Riscul de sangerare este proportional cu gradul trombocitozei, alterarea functiei plachetare, timpul de sangerare. Tratamentul antiagregant plachetar la pacientii cu trombocitoza poate creste riscul hemoragic.
factori de risc : virsta peste 70 ani, antecedente vasculare, tratamentul prin sangerari.
v Complicatii hematologice
evolutia spre mielofibroza cu metaplazie mieloida hepatosplenica. Este semnalata la 3 - 10 % din pacienti. Survine dupa 8-10 ani de evolutie. Aparitia este mai precoce la cei tratati de fond, prin sangerari. Se manifesta prin cresterea volumului splinei, evolutie spre anemie cu eritrocite deformate (hematii in lacrima - dacriocite), mielemie si eritroblasti in sangele periferic. La biopsia medulara se evidentiaza o fibroza colagenica intensa, mutilanta. Ulterior poate evolua spre leucemie acuta. Durata mediana de supravietuire dupa instalare, este de 2 ani.
transformare in leucemie acuta. Survine in 15-20% din cazuri, fie direct, fie precedat de osteomielofibroza (50% din cazuri). Transformarea este favorizata de tratamentul chimioterapic (fosfor radioactiv, busulfan, clorambucil). In majoritatea cazurilor, se transforma in leucemii mieloblastice, mai rar in leucemii limfoblastice, mielodisplazii sau leucemie mielo-monocitara cronica.
alte neoplazii - pacientii tratati cu Clorambucil sau fosfor radioactiv au o incidenta crescuta a cancerelor de piele si tumorilor gastrointestinale.
v Ulcerul peptic - frecventa bolii ulceroase este de 3 - 5 ori mai mare decat in populatia generala.
v Sindromul Budd-Chiari - apare la pcientii cu boli mieloproliferative, mai frecvent la sexul feminin. Se datoreaza trombozelor cu obstructie venoasa in teritoriul venei cave inferioare si a venei porte antrenand ascita, hepatosplenomegalie, dureri abdominale, hemoragii digestive superioare. Aproape 20% dintre pacienti pot fi asimptomatici. Diagnosticul se stabileste prin examene echografice, CT, RMN si/sau angiografie. Rezolvarea poate fi chirurgicala - sunturi sau transplant hepatic.
Evolutia bolii, in absenta tratamentului este spre deces intr-un interval relativ scurt. Astfel, 50% dintre pacientii netratati, decedeaza in primele 18 luni de la primul simptom. Principala cauza este tromboza. Dintre pacientii tratati exclusiv prin flebotomii, 50% decedeaza in primii 3,5 ani, iar dintre cei tratati cu citostatice vor deceda in intervalul intre 7 si 13 ani.
Scopul tratamentului in policitemia vera este de a controla numarul de celule circulante, minimaliza riscul complicatiilor legate de numarul crescut de celule, respectiv tromboza, de a diminua organomegalia simptomatica, de a controla pruritul. Tratamentul mielosupresor pare cel mai efectiv.
a) Metodele terapeutice :
1. Flebotomii (Singerari)
permit reducerea rapida a hipervolemiei si hipervascozitattii sanguine, astfel constituie tratamentul de urgenta al situatiilor amenintatoare.
induc hiposideremie cu diminuarea secundara a sintezei de hemoglobina si deci, a masei globulare totale.
avantaje : usor de practicat, nu necesita tehnica sau instrumentar sofisticat putandu-se utiliza rapid si oriunde, nu favorizeaza evolutia spre leucemii acute.
dezavantaje : necesita deplasari repetate din partea bolnavului, nu controleaza leucocitoza si nici hiperplachetoza, hiposideremia secundara determina astenie si stimularea formarii de trombocite (cu cresterea riscului trombotic), favorizeaza evolutia spre osteomielofibroza.
tratamentul de atac se face cu sangerari de 300-400 ml la doua zile interval pana la normalizarea hematocritului (Ht<48%) apoi ritmul este dictat de valoarea Ht (in general la 2-3 luni).
la persoanele in varsta sau la cei cu afectiuni vasculare, la care sangerarea ar putea antrena un dezechilibru volemic, dupa sangerare se perfuzeaza un volum corespunzator de plasma sau o solutie macromoleculara (Dextran) pentru reechilibrare.
studiul curbelor de supravietuire la pacientii cu policitemie a aratat ca supravietuirea cea mai lunga o au cei tratati exclusiv prin sangerare, comparativ cu celelalte tipuri de tratamente. Totusi, pentru o parte din pacienti, sangerarile repetate, pe termen lung sunt rau tolerate, fie in plan psihologic, fie datorita aparitiei complicatiilor : cardiovasculare (trombotice), cresterea progresiva a numarului de trombocite, peste 800.000-1.000.000/mm3, cresterea progresiva a volumului splenic, cu dezvoltarea unei metaplazii mieloide. Aceste situatii impun oprirea sangerarilor, cu schimbarea terapiei.
2. Fosfor radioactiv (P32)
introdus de Lawrence, a fost primul agent mielosupresor utilizat.
dupa injectare el se concentreaza in tesutul hematopoietic, ficat, splina si actioneaza local. Se incorporeaza in celulele care se divid activ, iar pe unele dintre ele, le iradiaza letal.
se administreaza 0,1 mCi/kg sau 2-3 mCi/m2 in doza unica. Granulocitele si plachetele raspund imediat, raspunsul fiind complet la trei saptamani de la tratament. Durata de viata a eritrocitelor fiind mai lunga, eficacitatea terapeutica nu poate fi apreciata corect inainte de doua - trei luni de la tratament. In acest interval, daca exista riscul de hipervascozitate se pot efectua sangerari, dar numai dupa trei saptamani de la tratament. Daca raspunsul este insuficient, la trei luni se mai poate administra o doza de 1 - 4 mCi.
remisiunea survine la 75-90% din cazuri si are o durata mediana de 22 luni. Doza recomandata trebuie respectata, ea putand fi redusa cu o treime la persoane peste 70 ani cu rezerve hematologice reduse.
in caz de reevolutie a bolii, dupa perioada de remisiune, se poate administra din nou fosfor radioactiv, in aceleasi doze, dar durata de remisiune, ca si procentul de remisiuni obtinute, scad progresiv, mai ales dupa a patra cura.
este un produs usor de manevrat, cu toleranta buna ce nu necesita o supraveghere stricta dar favorizeaza evolutia spre o leucemie acuta. Aceasta apare la aproximativ 15% din cei tratati, dupa un interval de 2 pina la 8 ani de la tratament.
in prezent indicatiile acestui tip de tratament raman cazurile de :
pacienti la care hidroxiureea este rau tolerata sau ineficienta
pacienti care refuza supravegherea stricta sau sa urmeze un tratament in mod regulat
pacienti in varsta, cu factori de risc trombotic.
3. Chimioterapie
Busulfan 4-6 mg/zi sau Clorambucil 4-10 mg/zi - sunt agenti alkilanti. Raspunsul maxim apare dupa 2 - 4 luni de tratament. Se poate instala, uneori, o remisiune de durata (pana la cativa ani). Se folosesc, insa, foarte rar datorita mielotoxicitatii crescute cu aplazie medulara, mai ales trombopenie, si favorizarea transformarii in leucemie acuta.
Hidoxiuree : reprezinta un puternic citostatic nealkilant care inhiba proliferarea celulara prin scaderea sintezei de ADN datorita inhibitiei ribonucleotid difosfat reductazei. A devenit drogul mielosupresiv cel mai folosit la pacientii cu policitemia vera. Se administreaza in doze de 1-2 g/zi sau 14-20 mg/kg/zi sub contol periodic al hemogramei. Este eficace pe toate cele trei linii mai ales pe trombocite, si este indicata in cazurile cu trombocitoza. Este eficace, obtinandu-se remisiuni la 90-95% din pacienti dupa 2-3 luni de tratament. Remisiunile pot fi de lunga durata, dar necesita tratament de intretinere in doze de 7,5 mg/kg/zi. Se poate asocia cu sangerari marind eficacitatea si reducand riscul trombotic. Riscul leucemogen este minor.
Pipobroman (Vericyte) este un agent alkilant, derivat de piperazina, eficace in tratamentul poliglobuliei. Se administreaza in doze de 1 mg/kg/zi apoi intretinere cu 0,1-0,2 mg/kg/zi. Ca si hidroxiureea, determina tulburari digestive, leuco si trombopenie.
Anagrelid - reprezinta un derivat de quinazolina creat initial ca inhibitor al functiei plachetare. Administrat in doze de 0,5-1 mg x 4/zi, per os antreneaza scaderea trombocitelor sub 600.000/mm3 in 2-4 saptamani la peste 90% de pacienti. Nu afecteaza linia granulocitara. Ca efecte secundare se semnaleaza edeme, vazodilatatie, insuficienta renala, insuficienta cardiaca manifesta.
Interferon - este utilizat datorita activitatii sale mielosupresive si datorita capacitatii de a antagoniza actiunea a factorului de crestere plachetar produs de megakariocite cu rol de a stimula proliferarea fibroblastilor. S-a demonstrat relativ recent eficacitatea sa in controlul mieloproliferarii si reducerea incidentei accidentelor tromboembolice. Se recomanda initierea unui tratament cu doze de 3MUI x 3/saptamana pentru a scadea dozele din al doilea an de utilizare (1-2MUI x 3/saptamana). Noi studii sunt necesare pentru aprecierea eficacitatii pe termen lung.
Tabelul IV
|
Trata ment |
SAnger|ri |
Fosfor (P32) |
Hidroxi uree |
Pipobro man |
Utilizare |
250-300 ml x 3/sapt pana ce Ht < 50% apoi la 2-3 luni |
0,1 mCi/kg sau 2 -3 mCi/m2 +/- repetare la trei luni |
1 - 2 g/zi sau 14-20 mg/kg/zi apoi 7,5 mg/kg/zi |
1 mg/kg/zi apoi 0,1-0,2 mg/kg/zi |
Indicatii |
Ø Pacienti tineri fara fac- tori de risc trombotic Ø Femei ce vor copii |
Ø Virstnici cu risc vascular Ø Hidroxiuree ineficace Ø Refuz tratament continuu |
Pacienti sub 65-70 ani cu factori de risc vascular |
|
|
Complicatii |
Ø Impact psihologic Ø Hiposideremie Ø Hiperplachetoza Ø Risc trombotic Ø Metaplazie mieloida |
Ø Leucemie acuta Ø Sindrom mielodisplazic |
Tulburari digestive Trombocitopenie Leucopenie |
|
|
Rezistenta |
P32 sau citostatice in functie de varsta |
Pipobroman Hidroxiuree |
Interschimbare Fosfor radioactiv Busulfan |
|
b) Tratament complementar
in caz prurit se incearca, cu rezultate variabile : ciproheptadina, colestiramina, cimetidina
in caz de deficit martial : se recomanda tratamentul cu fier. S-a observat faptul ca eritrocitele microcitare poseda o vascozitate intrinseca mai mare la oricare nivel al hematocritului, putand favoriza complicatiile. Astfel se recomanda evitarea microcitozelor severe prin sangerari multiple. Atunci cand CHEM este sub 22 pg se recomanda aportul medicamentos de fier, prudent, sub monitorizarea Hb si Ht.
in caz hiperuricemie sau crize de guta : allopurinol : 200-300 mg/zi.
in caz de splenomegalie masiva se poate incerca administrarea de androgeni sau citostatice. Hydrea sau IFN pot antrena diminuarea splinei. Iradierea splenica in doze mici poate fi eficace. S-a incercat splenectomia dar s-a observat, adesea, o evolutie spre hepatomegalie compensatorie progresiva si chiar transformarea in leucemie acuta.
c) Strategia terapeutica - principii directoare - nu intruneste un consens unanim.
Terapia trebuie adaptata fiecarui caz in parte, tinand cont de status-ul general, cellular si al organomegaliilor.
Indiferent de varsta pacientului, prima urgenta o constituie scaderea Ht in limite de sigutanta (sub 48%), cu atat mai mult la pacientii simptomatici. Deci, la toti pacientii simptomatici si cu valori cu risc crescut, se va debuta tratamentul prin flebotomii. Tratamentul de atac se face cu sangerari de 300-400 ml la doua zile interval (250 ml la 3 zile la pacientii in varsta si cu afectiuni cardiovasculare manifeste) pana la normalizarea hematocritului (Ht<48%) apoi ritmul este dictat de valoarea Ht (in general la 2-3 luni). Continuarea tratamentului se face in functie de varsta si factorii de risc vascular.
Se recomanda suprimarea miloproliferarii cu ajutorul chimioterapiei - (HU) la toti pacientii peste 50 ani. In general, 32P trebuie rezervat pacientilor peste 80 ani sau celor cu patologii associate la care speranta de viata este sub 5-10 ani, dat fiind riscul de leucemizare la 10-15 ani. Pacientii cu tendinta la tromboze sau care dezvolta trombocitoza post- flebotomie trebuie tratati chimioterapic. Anagrelidul trebuie rezervat pacientilor de 50-70 ani.
Valorile globulare vor fi mentinute in limite de siguranta prin control periodic si adaptare therapeutica
Se va evita supratratarea si toxicitatea prin utilizarea judiciousa a chimioterapiei si radioterapiei. Se va prefera suplimentarea flebotomiilor utilizarii in exces a mielosupresoarelor.
Se va temporiza orice interventie chirurgicala pana la stabilizarea bolii (risc hemoragic sau trombotic crescut).
Femeile in periada de procreiere vor fi tratate prin flebotomii. La pacientii de sex masculine, tineri, mielosupresia poate duce la aspermie - astfel evaluati bine cazul inainte de a recurge la chimio sau radioterapie.
Pentru pacientii cu risc trombotic se poate asocia tratament cu antiagregante plachetare : Aspirina 50-100 mg/zi si/sau Dipiridamol 75 mg x 3/zi
I. Poliglobuliile false - sunt situatii in care masa eritrocitara totala nu este crescuta in mod real, ci doar relativ, in raport cu volumul plasmatic care este scazut prin pierderi. Astfel de situatii apar in deshidratari masive (varsaturi abundente, diarei profuze, transpiratii profuze, tratament diuretic nesupravegheat), si in arsuri intinse. In cazul talasemiilor heterozigote putem evidentia o policitemie asociata cu microcitoza si discreta anemie.
II. Poliglobuliile secundare se definesc prin cresterea masei globulare totale datorate unei cresteri a secretiei de eritropoietina. Hipersecretia poate fi secundara unei hipoxii tisulare sau independenta :
A. Cu hipoxie tisulara
v Prin scaderea saturatiei in oxigen a sangelui arterial (PaO2 < 65 mmHg, SaO2<92%)
Boli pulmonare - BPOC, intoxicatie cronica cu CO - hipoxemia din aceste boli este asociata cu eritrocitoza, existand o corelatie direct proportionala intre cele doua. Volumul plasmatic este adesea crescut incat, adesea, Hb si Ht sunt subestimate, ca si severitatea eritrocitozei. Pacientii cu Ht peste 55 % au indicatie sangerare, cu atat mai mult cei care asociaza semne de decompensare cardiaca.
Hipoventilatie alveolara (ex boala Pickwick) - poate asocia poliglobulie. In aceste cazuri hipoxia poate sa nu fie aparenta la analizele practicate uzual, in cursul zilei. Se impune determinarea gazelor arteriale la pacientul adormit cand survin episoadele de apnee.
Boli cardio-vasculare - pacientii cu cardiopatii congenitale de tip comunicare dreapta-stanga pot prezenta cresteri ale Ht. Pentru unii din ei cresterea poate fi progresiva cu cresterea vascozitatii sanguine sI afectarea capacitatii de transport a oxigenului.
Tabagism - cauza frecventa a unei poliglobulii discrete asociind o polinucleoza moderata. La fumatorii cronici, oxidul de carbon rezultat din fumat se fixeaza cu afinitate crescuta pe hemoglobina formand carboxihemoglobina. In consecinta scade saturatia sangelui arterial in oxigen cu stimularea secundara a secretiei de eritropoietina. Rezulta o productie crescuta pe seria rotie cu poliglobulie ce va creste vascozitatea sanguina cu afectarea secundara a transportului de oxigen.
Sejur prelungit la altitudine - ascensiuni la altitudini peste 2 000 - 2 500 m antreneaza initial o scadere cu 20 % a volumului plasmatic cu poliglobulie falsa (relativa) pentru ca ulterior sa fie urmata de o crestere a secretiei de eritropoietina cu poliglobulie adevarata.
v Fara desaturare in oxigen a sangelui arterial :
Hemoglobine anormale cu afinitate crescuta pentru oxigen
Methemoglobinemie secundara sau congenitala (in contextul hemoglobinopatiilor)
Deficit in 2,3-DPG
Metode de diagnostic : masurarea gazelor sanguine, determinarea carboxi-hemoglobinei, electroforeza hemoglobinei, dozare 2,3-DPG, dozarea eritropoietinei (crescuta).
B. Fara hipoxie tisulara :
v Tumori secretante de eritropoietina sau substante cu activitate eritropoietinica - exista multiple tipuri de tumori cu diferite localizari capabile sa secrete eritropoietina sau alte substante cu activitate eritropoietinica. Aceasta secretie ectopica poate antrena cresteri ale masei eritrocitare de pana la doua ori nivelul normal. Pentru unele tumori se asociaza si alte acuze si semne ce permit diagnosticul. Pentru altele aceasta poliglobulie paraneoplazica poate constitui singurul semn precoce ce ar permite diagnosticarea si eradicarea tumorii intr-un stadiu curabil. Tumori secretante :
Carcinom renal
Carcinom hepatocelular
Hemangiom hepatic
Hemangioblastom cerebelos
Leiomiom uterin
Carcinom de ovar
Feocromocitom
Altele
v Leziuni renale netumorale : hidronefroza, chisturi renale, adenoame, ischemie renala prin stenoza de artera renala;
v Endocrine : Boala Cushing, feocromocitom, terapie prelungita cu hormoni androgeni.
Metode de diagnostic : gazele arteriale, echografie abdominala, urografie, scanner abdomino-pelvin, cerebral, dozarea eritropoietinei (crescuta).
C. Poliglobulii ereditare (secundare sau primitive)
v Hemoglobinopatii cu afinitate crescuta pentru oxigen
v Scaderea 2,3-difosfogliceratului eritrocitar
deficit in 2,3-DPG mutaza
cresterea ereditara a adenozin-trifosfatului eritrocitar
v Eritrocitoza esentiala - productie crescuta de eritropoietina
v Polycytemia vera familiala si congenitala (mecanism poligenic) - boala cu transmitere autozomal dominanta, caracterizata prin masa eritrocitara crescuta, nivel crescut al Hb, hipersensibilitatea progenitorilor eritrocitari la EPO, nivel seric scazut al EPO, curba de disociere Hb-O2 normala si absenta progresiei spre leucemie. Mutatia poate sa apara la nivelul genei receptorului pentru EPO (EpoR) sau la nivelul genelor altor structuri implicate in calea te transmitere a semnalului proliferant la nivelul progenitorilor eritocitari cu productie excesiva, neinhibata.
III.Poliglobulia aparenta - numita si sindrom Gaisbck
pacientii au un Ht crescut dar masurarea masei eritocitare totale da valori normale. Aspectul rezulta din asocierea unei mase eritrocitare totele la limita superioara a norma lului cu un volum plasmatic la limita inferioara a normalului sau chiar scazuta.
Mecanismul ramane necunoscut dar par a juca un oarecare rol : fumatul, hipertensiune, consumul cronic de alcool
Boala survine predominant la pacienti de sex masculin, adesea moderat obezi, cu un profil anxios. Majoritatea sunt fumatori (atentie la poliglobulia secundara la fumatori).
Boala se manifesta prin astenie, fatigabilitate, cefalee, ameteli, dispnee de effort, jena epigastrica.
Examenul clinic evidentiaza : eritroza tegumentara si conjunctivala, hepato-splenomegalie (foarte rar), HTA (frecvent).
Laborator : Ht moderat crescut, GA, Tr, Reticulocitele sunt normale, maduva osoasa are aspect normal
Tratament : se recomanda sistarea fumatului si a consumului de alcool, controlul greutatii si al valorilor tensionale. Sangerarile se recomanda la cei cu persistenta valorilor crescute ale Ht dupa adoptarea masurilor precedente, si cu semne datorate hipervascozitatii sanguine.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 14284
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved