| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
Bolile renale chistice
Chisturile apar prin dilatatia unei portiuni din nefron cu formarea unei cavitati acoperite cu epiteliu, pline cu lichid sau material semisolid. La formarea chisturilor pot interveni: leziuni ale matricei extracelulare inconjuratoare, secretie continua de lichid, hiperplazia epiteliului din peretele chistic. [
Clasificare
1. Boli ereditare
A. Afectare cortico-medulara
a. Transmitere autozomal dominanta
-Boala polichistica renala autozomal dominanta
-Scleroza tuberoasa
-Boala von Hippel-Lindau (descrisa in subcapitolul "Afectarea renala in facomatoze")
-Boala glomerulochistica
b. Transmitere autozomal recesiva
-Boala polichistica renala autozomal recesiva
B. Afectare medulara
-Boala chistica a medularei
2. Boli non-ereditare
-Boala chistica hipokaliemica
-Chisturi simple
-Boala chistica renala dobandita
-Rinichiul spongios medular
-Rinichiul multichistic congenital [12]
Boala polichistica renala autozomal dominanta (ADPKD)
Este o afectiune sistemica caracterizata prin prezenta de chisturi distribuite difuz in ambii rinichi. Gena responsabila este localizata pe cromozomul 16 (tipul 1) sau pe cromozomul 4 (tipul 2).
Clinic, cele 2 forme sunt diferite: ADPKD tipul 1 debuteaza mai precoce, iar HTA si BCR apar mai devreme decat in tipul 2. Boala afecteaza ambele sexe in aceeasi masura, manifestandu-se in decadele 3-4 de viata. [12, 13]
Anatomie patologica
Macroscopic rinichii pot atinge dimensiuni foarte mari (pana la
M.O evidentiaza chisturile ca diverticuli in capsula Bowman, tubul proximal, ansa Henle si tubul distal.
M.E a demonstrat ca, pe masura ce cresc, chisturile nu comunica cu tubii de origine.
Chisturile cu origine in nefronul distal contin fluid cu concentratie scazuta in Na si concentratii mari de protoni, K, uree, creatinina. Aceste chisturi nu concentreaza antibioticul. Restul chisturilor au concentratie de electroliti asemanatoare cu cea din ser si concentreaza bine antibioticul. [13]
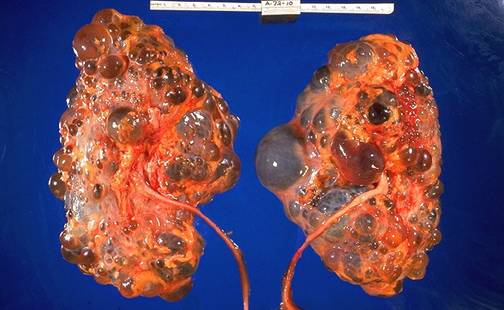
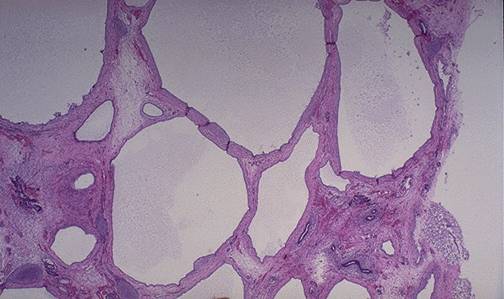
Figura 9.2. Aspect macroscopic si microscopic (Hematoxilina-eozina) in boala
polichistica hepato-renala autozomal dominanta. Adaptat dupa Cho ME, Kopp JB, Fabry disease in era of enzyme replacement therapy: a renal perspective. Pediatr Nephrol 2004, 583-593
Clinic
Manifestari renale
Exista o multitudine de semne si simptome aparute la debut, determinate de evolutia bolii sau de aparitia complicatiilor. Exista insa si pacienti care pot pot fi asimptomatici pana in stadiul avansat al BCR.
Durerea abdominala este simptomul cel mai frecvent . Poate fi determinata de intinderea capsulei si tractiunea pediculului renal ( cronic) sau de ruptura unui chist, litiaza sau neoplasm (acut).
Poliuria apare in fazele avansate ale bolii. HTA apare, insa, precoce la mare parte din pacienti, fiind determinata de marirea chisturilor si activarea sistemului renina-angiotensina-aldosteron.
Hematuria poate fi o modalitate de debut frecvent intalnita si cauzata fie de ruptura chisturilor, fie de nefrolitiaza si infectie. Atunci cand apare dupa varsta de 50 de ani si este asociata cu febra si scadere ponderala se poate suspiciona un neoplasm renal.
Litiaza renala e prezenta la aproximativ 1/5 cazuri. Pot aparea infectii ale tractului urinar inferior dar si pielonefrite. Pielonefritele evolueaza cu piurie, cilindrii leucocitari, uroculturi pozitive. Lipsa de raspuns la terapie dupa 3-4 zile sugereaza prezenta de piochist. [12, 11]
Manifestari extrarenale
Chisturile hepatice nu afecteza functia hepatica, dar compresiunea la nivelul parenchimului hepatic poate necesita interventie chirurgicala.
Chisturile pancreatice sunt intalnite la aproximativ 10% din pacienti si 5% din pacienti au chisturi splenice.
Diverticulii colonici apar cel mai frecvent la pacientii hemodializati.
Afectare cardiaca: prolaps de valva mitrala, insuficienta aortica, HVS secundara HTA.
Anevrismele intracraniene (1-40%) au risc de ruptura mai mare decat la populatia generala. [11]
Evolutie. Prognostic
Caracteristic pentru evolutia bolii este discrepanta dintre gradul anemiei (usoara) si nivelul crescut al creatininemiei.
Peste jumatate din pacienti evolueaza spre BCR, barbatii mai precoce decat femeile. Chisturile hepatice evolueaza de cele mai multe ori benign. In cazuri cu infectii severe, recurente, hematurie severa, recurenta sau in cazul suspiciunii unui carcinom renal poate fi necesara nefrectomia.
La pacientele cu afectiune necomplicata evolutia sarcinii este normala si sarcina nu agraveaza boala.
Pacientii cu ADPKD tolereaza bine HD, dar exista un risc crescut al complicatiilor trombotice. Dializa peritoneala este formal contraindicata la pacientii cu dimensiuni foarte mari ale chisturilor datorita disconfortului abdominal produs de lichidul de dializa, a complicatiilor infectioase mai frecvente si a complicatiilor hemoragice.
Transplantul renal evolueaza favorabil. [12]
Tratament
Tratamentul este simptomatic si/sau al complicatiilor.
Durerea: analgezicele pot fi utilizate. Pentru chisturile gigante se poate realiza drenajul percutan, scleroza cu etanol sau tratament chirurgical (aspirarea fluidului si indepartarea chirurgicala a peretului chistic).
HTA: Se administreaza diuretice, cu prudenta IECA. Se mai pot utiliza Ca-blocantele, beta-blocantele.
Anevrismele cerebrale: riscul ruperii lor este maxim la peste 1cm. Peste aceasta dimensiune se poate lua in discutie interventia chirurgicala.
Hematuria: necesita spitalizare; in absenta infectiei este suficient repausul la pat minimum 48 ore, tratamentul HTA si asigurarea unei diureze peste 2000 ml/zi.
Infectia urinara: poate fi infectie urinara joasa, PNA sau infectia chisturilor. PNA se trateaza cu antibiotice administrate parenteral ulterior per os pana cand piuria si simptomele se remit timp de doua saptamani. Absenta raspunsului la antibioticele lipofobe este sugestiva pentru infectia chisturilor. In acest caz, se administreaza antibiotice liposolubile (ciprofloxacina, norfloxacin) timp de 3-6 luni.
Litiaza renala: este frecvent cu continut de oxalat de calciu. Este necesara interventia urologica in cazul obstructiei complicata cu infectie severa cu germeni rezistenti. [12, 13]
Boala polichistica renala autozomal recesiva (RDPKD)
Afectiunea se caracterizeaza prin prezenta de chisturi renale bilaterale fara displazie, ficatul fiind afectat prin fibroza. Boala se transmite autozomal recesiv, gena responsabila fiind localizata pe cromozomul 6. Este evidenta la nastere si frecvent letala in primul an de viata.[15]
Anatomie patologica
La nastere rinichi sunt mariti de volum, dimensiunile sunt maxime intre 1 si 2 ani, incepand sa scada pana la 4-5 ani. Chisturile se dezvolta din portiunea terminala a tubilor colectori. Sunt structuri aliNefropatii interstitiale acutete si dilatate radial. Ectazia ductelor medulare este patognomonica. Ficatul prezinta fibroza portala, cu ducte biliare dilatate. In evolutie se dezvolta hepatosplenomegalie si hipertensiune portala. [15]
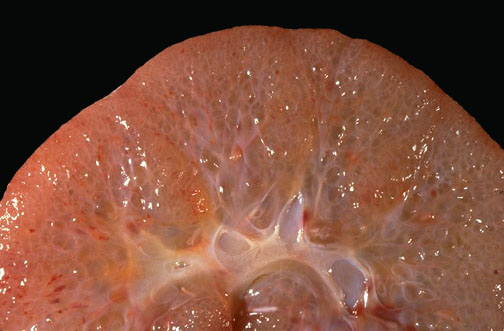
Figura 9.3. Aspect macroscopic al rinichiului in RDPKD, adaptat dupa The University of Utah Eccles Health Sciences Library
Clinic
Nou-nascutii pot avea facies Potter cu fante oculare adinci, nas aplatizat, micrognatie, urechi mari. Majoritatea decedeaza prin sindrom de detresa respiratorie, determinata de hipoplazie pulmonara, sau prin pneumotorax. Cei diagnosticati dupa primul an de viata prezinta greata, varsaturi, dureri abdominale cu hepatosplenomegalie. Majoritatea copiilor prezinta HTA. Pe masura ce dezvolta BCR apar tulburari de crestere, anemie, osteodistrofie renala.[15]
Diagnostic pozitiv si diferential
Diagnosticul pozitiv se stabileste pe
istoricul familial, examenul ecografic al tuturor membrilor familiei, rar
biopsie hepatica. Ecografia
efectuata intrauterin sau neonatal evidentiaza rinichi cu diminsiuni crescute
cu margini imprecis delimitate. Rar apar chisturi renale de obicei sub
Diagnosticul diferential se face cu tumora Wilms bilaterala, boala glomerulochistica, ADPKD.[15]
Evolutie
Copii diagnosticati in primul an de viata au mortalitate mare (60%). De obicei nou-nascutul este anuric si cu insuficienta respiratorie. Cazurile rare care supravietuiesc perioadei neonatale evolueaza progresiv spre BCR dupa aproximativ 5 ani. Sindromul de insuficienta hepatica apare dupa aproximativ 5 ani de la nastere si e insotit de hipertensiune portala cu hemoragie digestiva superioara frecventa si hipersplenism hematologic.[16]
Tratament
Tratamentul este simptomatic. In cazul initierii HD se recomanda un aport proteic de 1,5-2 g/kgcorp/zi. HD nu este contraindicata, dar avand in vedere existenta fibrozei hepatice progresive este recomandat dublu transplant hepatic si renal. [15]
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3473
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved